
They're different. Confusing them causes real problems: failed audits, 510(k) submissions sent back for insufficient biocompatibility evidence, or materials selected on the wrong basis for patient-contact applications.
This article breaks down what each standard actually tests, where they overlap, and how to determine which one your rubber seal application requires.
Key Takeaways
- USP Class VI uses three animal-based in vivo tests to confirm a rubber compound isn't acutely toxic — the baseline standard in pharmaceutical and biotech fluid systems
- ISO 10993 is a 20+ part framework that assigns a risk-based test matrix based on contact type, location, and duration; required for FDA 510(k) and PMA submissions
- Both can apply to rubber seals, but they test different things and are not interchangeable
- For FDA device submissions, ISO 10993-1 is the organizing framework; USP Class VI data is supplementary, not a substitute
- Your application type, contact duration, and target markets determine which standard — or both — you need
USP Class VI vs ISO 10993: Quick Comparison
These two standards differ in scope, application, and regulatory weight. Here's how they compare across the factors that matter most for seal material selection:
| Factor | USP Class VI | ISO 10993 |
|---|---|---|
| Testing scope | Fixed: 3 in vivo biological reactivity tests | Risk-based: test matrix varies by contact type, duration, and body location |
| Governing body | U.S. Pharmacopeia (non-governmental) | International Organization for Standardization |
| FDA recognition | Accepted in pharmaceutical/drug-contact contexts; not a standalone for device submissions | Directly referenced in FDA guidance for 510(k), PMA, and De Novo submissions |
| Best fit | Pharmaceutical manufacturing, biotech fluid systems, drug-contact seals | FDA-regulated medical devices, implantables, global market submissions |
| Cost & timeline | Lower cost, faster — three defined tests | Broader scope, longer timeline — matrix scales with device risk |
What Is USP Class VI Certification?
The Three Required Tests
USP Class VI is the highest biocompatibility category within the U.S. Pharmacopeia's plastics classification system, governed by USP General Chapter <88> Biological Reactivity Tests, In Vivo. For a rubber compound to achieve Class VI status, it must pass three specific in vivo tests:
- Systemic Injection Test — Material extract is injected into mice and monitored for systemic toxic response
- Intracutaneous Test — Extract is injected into rabbit skin to detect local tissue reactions
- Implantation Test — Material samples are implanted into rabbit muscle tissue and evaluated for localized tissue response
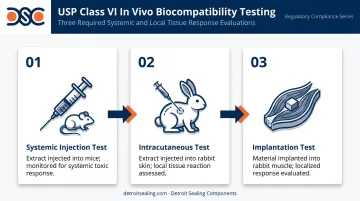
All three tests assess the material's extractables — substances that leach out of the rubber compound under defined conditions. The certification establishes that those extractables don't produce harmful biological reactions.
Why It Matters for Rubber Seals
Seals in pharmaceutical manufacturing lines, IV systems, drug delivery components, and biotech processing equipment come into direct contact with drugs, biologics, or bodily fluids. USP Class VI certification confirms the rubber compound won't contaminate those media with harmful leachables.
The rubber families most commonly certified to USP Class VI include:
- Silicone (VMQ/LSR) — most prevalent; platinum-cure formulations preferred for lower extractables
- EPDM — common in pharmaceutical fluid systems
- FFKM (perfluoroelastomers) — used in aggressive chemical environments
- FKM — select formulations only
Certification is compound-specific, not material-category-specific. Not all silicone qualifies — the exact formulation, cure system, and processing must each be tested and documented. A generic claim that "our silicone is USP Class VI" without compound-specific test data is not sufficient for sourcing decisions.
The Key Limitation
USP Class VI tests a fixed set of biological endpoints regardless of how the device actually contacts the body. It doesn't cover genotoxicity, chronic toxicity, hemocompatibility, or carcinogenicity — endpoints that may be required for long-term or patient-contact device applications.
Pacific BioLabs notes that USP class plastics testing is not a substitute for comprehensive medical device biocompatibility evaluation. USP has also signaled revisions to <88> with a targeted effective date of December 2026 — meaning compliance requirements for new material approvals may shift before that date.
For rubber seal sourcing, DSC's ISO 17025 accredited lab capability supports compound-level verification — confirming whether a specific formulation's existing Class VI data actually covers the contact conditions and extractables profile of your application.
What Is ISO 10993 Biocompatibility Testing?
The Risk-Based Framework
ISO 10993 is a series of international standards that establishes a risk-based approach to medical device biocompatibility evaluation. Unlike USP Class VI's fixed three-test package, ISO 10993 requires manufacturers to build a testing matrix tailored to the specific device.
The foundational document, ISO 10993-1, defines how to categorize devices by two dimensions:
Contact type:
- Surface contact (intact skin, breached skin, mucosal membrane)
- External communicating (blood path indirect, tissue/bone/dentin, circulating blood)
- Implant (tissue/bone, blood)
Contact duration:
- Limited: under 24 hours
- Prolonged: 24 hours to 30 days
- Permanent: over 30 days
As contact intimacy and duration increase, the required test matrix expands. A seal in an external diagnostic device needs far less testing than a seal in a long-term implantable device.

Key Standards in the Series
The ISO 10993 series covers 20+ individual standards. The most relevant for rubber seals:
- ISO 10993-5 — In vitro cytotoxicity (nearly universal baseline)
- ISO 10993-10 — Sensitization
- ISO 10993-3 — Genotoxicity, carcinogenicity, reproductive toxicity
- ISO 10993-4 — Hemocompatibility (required for blood-contacting seals)
FDA's Current Position
FDA issued its final guidance on ISO 10993-1 in September 2023, covering PMA, HDE, IDE, 510(k), and De Novo submissions. ISO 10993-1 is the organizing framework for device biocompatibility submissions.
FDA evaluates the finished medical device — not individual materials. USP Class VI data can be submitted as supporting information, but it doesn't substitute for an ISO 10993-based biocompatibility evaluation in a formal device submission.
For device OEMs, ISO 10993 offers a practical advantage beyond FDA compliance. Because the framework is internationally harmonized, a single compliant dossier covers multiple regulatory bodies:
- FDA (United States)
- EU Notified Bodies (CE marking under MDR)
- Health Canada (Canadian device submissions)
Which Standard Does Your Application Actually Require?
The Decision Framework
The primary driver isn't personal preference — it's your regulatory pathway and application context. Three scenarios cover most situations:
Scenario 1 — Pharmaceutical / biotech fluid systems: USP Class VI is typically the baseline expectation. Drug manufacturing equipment, bioprocessing seals, pharmaceutical tubing, and IV system components fall here. Formal FDA device clearance isn't required, but drug-contact safety evidence is.
Scenario 2 — FDA-regulated medical device submissions: ISO 10993 evaluation is required. USP Class VI data may support the compound qualification but won't satisfy the biocompatibility section of a 510(k) or PMA on its own.
Scenario 3 — Implantable or long-term patient-contact devices: ISO 10993 with an expanded endpoint matrix. FDA's biocompatibility endpoint table for long-term/permanent implants includes genotoxicity, chronic toxicity, carcinogenicity, and hemocompatibility — none of which USP Class VI addresses.

Can a Seal Carry Both?
Yes — and for many applications, it should. Many high-quality medical-grade rubber compounds, particularly silicone and FFKM formulations, carry USP Class VI certification and have ISO 10993 data packages available. For seal distributors, offering compounds with dual documentation shortens customer qualification timelines considerably.
A Concrete Example
Take a silicone O-ring specified for a drug-delivery infusion pump. The OEM needs:
- USP Class VI data to satisfy drug-contact requirements for the pharmaceutical fluid path
- ISO 10993 cytotoxicity and sensitization data to satisfy the biocompatibility section of the 510(k) submission for the device itself
These standards aren't redundant — they address different aspects of the same component's safety profile. DSC maintains compound-level USP Class VI and ISO 10993 documentation across silicone, FFKM, and other medical-grade formulations. Procurement teams can confirm both certifications for a given compound in one conversation, rather than coordinating documentation across multiple suppliers.
Conclusion
USP Class VI and ISO 10993 aren't competing standards — they address different regulatory questions about the same rubber compound. USP Class VI establishes whether a material is acutely non-toxic based on standardized biological reactivity tests. ISO 10993 evaluates whether a device material poses unacceptable biological risks given its specific contact type, duration, and patient population.
For pharmaceutical manufacturing applications, USP Class VI is typically the right baseline. For any formal FDA device submission, ISO 10993-1 is the required framework — and USP Class VI data plays a supporting role, not a primary one.
Getting this right at the sourcing stage matters. Documentation gaps discovered during audits or FDA pre-submission reviews create costly holds. At the sourcing stage, verify:
- Material traceability to the specific rubber compound (not just material family)
- Compound-specific certification documentation (USP Class VI, ISO 10993 test reports)
- Biocompatibility data packages aligned to your regulatory submission pathway
Frequently Asked Questions
What materials are USP Class VI?
USP Class VI certification is compound-specific, not a blanket category approval. Commonly certified materials include silicone, EPDM, FFKM, and select FKM formulations — but the specific rubber compound formulation must be individually tested and certified. A generic material type claim without compound-level test documentation doesn't qualify.
What is the ISO 10993 series of standards?
ISO 10993 is a series of 20+ international standards governing the biological evaluation of medical devices. The foundational standard, ISO 10993-1, defines a risk-based framework for selecting biocompatibility tests based on device contact type (surface, communicating, implant) and duration (limited, prolonged, permanent).
Is rubber FDA approved?
Raw rubber materials are not subject to FDA approval. Specific compounds can be FDA compliant — conforming to regulations like 21 CFR 177.2600 for food-contact rubber or backed by biocompatibility data under ISO 10993 guidance. "FDA approved" is a designation that applies to finished medical devices, not component materials.
What does FDA compliant mean for rubber seals?
FDA compliant means a material meets the applicable FDA regulations and guidance documents for its intended use. For rubber seals, this typically involves conformance to 21 CFR extractables regulations and biocompatibility standards like ISO 10993. It does not mean FDA has reviewed or approved the specific seal.
How do I know if silicone is medical grade?
Medical-grade silicone is identified by documented biocompatibility certifications (USP Class VI or ISO 10993 data), platinum-cure chemistry rather than peroxide-cure, low extractables profiles, and full manufacturer traceability. Reputable suppliers will provide compound-specific test reports and certificates of conformance on request.
What is 21 CFR Part 820 for medical devices?
21 CFR Part 820 governs quality management systems for medical device manufacturers in the U.S. — now updated to align with ISO 13485, effective February 2026. It doesn't certify rubber materials directly, but requires manufacturers to document and verify component materials, including biocompatibility, as part of design and production controls.


